




À la différence des molécules d’hydrocarbures d’origine fossile, celles issues de la biomasse sont polaires, en raison des hétéro-atomes qu’elles renferment. Cette différence à l’échelle moléculaire induit un comportement macroscopique plus complexe dont il faut tenir compte pour le dimensionnement des procédés qui les mettent en œuvre.
En outre, si la variété des molécules rencontrées dans les fluides pétroliers est grande, elle l’est nettement plus dans la biomasse. Ceci rend impossible la réalisation de mesures sur chaque molécule ou sur chaque combinaison d’entre elles, en vue de caler les modèles thermodynamiques traditionnels. Disposer de moyens prédictifs pour calculer les propriétés de ces mélanges de manière purement théorique, à partir de la structure moléculaire, devenait dès lors une nécessité.
L’objectif de la chaire de recherche et d’enseignement « Thermodynamique pour les carburants issus de la biomasse » était la construction d’une telle méthode prédictive.
Pour cela, les chercheurs se sont appuyés sur des travaux académiques initiés dans les années 90 proposant des méthodes basées sur de la mécanique statistique pour construire des modèles thermodynamiques (équations d’état SAFT ).
Une liste non exhaustive des travaux menés au sein de la chaire, notamment au travers de thèses, permet d’illustrer les avancées obtenues :
- une méthode basée sur les contributions de groupes a été utilisée pour calculer les équilibres de phase pour des mélanges de molécules identifiées dans la biomasse[1]. Elle se concrétise par le modèle GC-PPC-SAFT dont le nom traduit le contenu[2,3] ;
- pour combler la limite du modèle vis-à-vis de molécules ayant de nombreuses fonctionnalités oxygénées, une méthodologie a été mise au point pour étendre les contributions de groupes en prenant en compte la distance entre groupes[4] ;
- pour certaines molécules de la biomasse aux formes particulièrement complexes, tel le guaiacol, la description moléculaire a dû être adaptée au solvant dans lequel la molécule se trouve (la figure en montre un exemple)[5] ;
- le modèle développé a aussi été confronté à des mesures de solvatation (soluté dans solvant) lors d’une collaboration avec une équipe de l’université de Kazan[6] ;
- GC-PPC-SAFT a également été utilisé dans le contexte des procédés d’estérification servant à la conversion des huiles végétales en carburant[7] ;
- enfin, pour dimensionner le procédé d’hydrotraitement des fluides issus de la biomasse, et les rendre ainsi compatibles avec les carburants pétroliers, la solubilité de l’hydrogène dans ces fluides a été étudiée[8].
Cliquer sur l'image pour l'agrandir
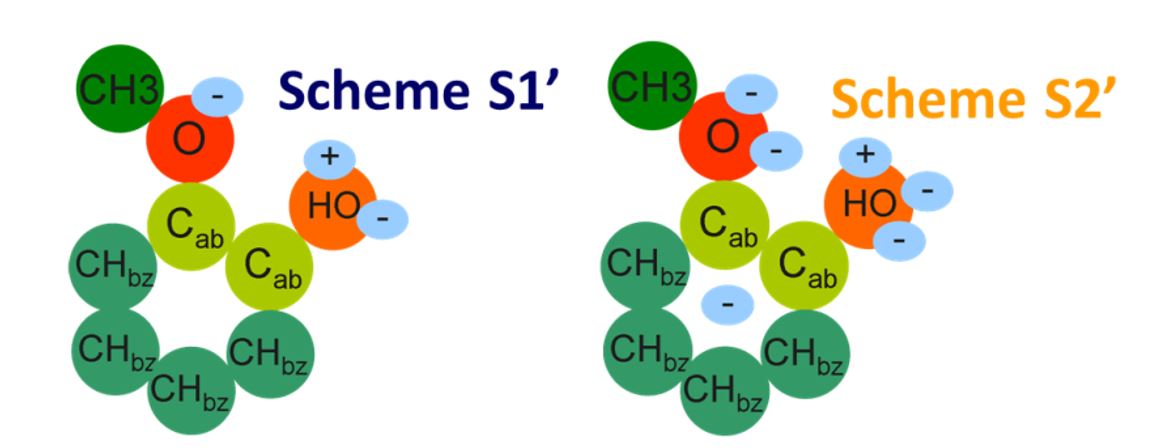
Plusieurs schémas de distribution de ces sites ont été étudiés.
Grâce aux travaux de cette chaire de recherche, IFPEN dispose aujourd’hui d’un outil de type « équation d’état », pour mieux comprendre et décrire les mélanges issus de la biomasse, avec leurs composés oxygénés. Il est désormais d’un usage courant dans des projets de recherche appliquée.
1- Chaire de la Fondation Tuck, 2009 à 2018.
2- Statistical Associating Fluid Theory.
3- Consistant à découper les molécules en fragments, bien moins nombreux que les molécules (exemple sur la figure) dont on considère que chacun contribue pour une partie à la propriété désirée.
4- GC pour les contributions de groupes et P parce qu’il comporte un terme polaire, tout en se basant sur la variante Perturbed Chain de SAFT
[1] D. Nguyen-Huynh, J.-C. de Hemptinne, R. Lugo, J.-P. Passarello, P. Tobaly, Simultaneous liquid–liquid and vapour–liquid equilibria predictions of selected oxygenated aromatic molecules in mixtures with alkanes, alcohols, water, using the polar GC-PC-SAFT (Prédictions simultanées d'équilibres liquide-liquide et vapeur-liquide de certaines molécules aromatiques oxygénées dans des mélanges avec des alcanes, des alcools et de l'eau, à l'aide du GC-PC-SAFT polaire), 92 (2014) 2912–2935.
>> https://www.sciencedirect.com/science/article/pii/S0263876214002470
[2] T.-B. Nguyen, J.-C. de Hemptinne, B. Creton, G.-M. Kontogeorgis, Characterization Scheme for Property Prediction of Fluid Fractions Originating from Biomass (Schéma de caractérisation pour la prédiction des propriétés des fractions fluides issues de la biomasse), ENERG FUEL 29 (2015) 7230–7241.
>> http://dx.doi.org/10.1021/acs.energyfuels.5b00782
[3] T.-B. Nguyen, J.-C. de Hemptinne, B. Creton, G.-M. Kontogeorgis, Improving GC-PPC-SAFT equation of state for LLE of hydrocarbons and oxygenated compounds with water (Amélioration de l'équation d'état GC-PPC-SAFT pour la LLE des hydrocarbures et des composés oxygénés avec de l'eau), Fluid Phase Equilib. 372 (2014) 113–125.
>> http://www.sciencedirect.com/science/article/pii/S0378381214002064
[4] M. Jaber, W. Babe, E. Sauer, J. Gross, R. Lugo, J.-C. de Hemptinne, An improved group contribution method for PC-SAFT applied to branched alkanes: Data analysis and parameterization (Une méthode de contribution de groupe améliorée pour PC-SAFT appliquée aux alcanes ramifiés: analyse et paramétrage des données), Fluid Phase Equilib. 473 (2018) 183–191.
>> https://www.sciencedirect.com/science/article/pii/S0378381218302504
[5] L. Grandjean, J.-C. de Hemptinne, R. Lugo, Application of GC-PPC-SAFT EoS to ammonia and its mixtures (Application de GC-PPC-SAFT EoS à l'ammoniac et ses mélanges), Fluid Phase Equilib. 367 (2014) 159–172.
>> http://dx.doi.org/10.1016/j.fluid.2014.01.025
[6] M.-A. Varfolomeev, R.-N. Nagrimanov, M.-A. Stolov, N. Ferrando, R. Lugo, J.-C. de Hemptinne, Guaiacol and its mixtures: New data and predictive models. Part 2: Gibbs energy of solvation (Guaiacol et ses mélanges: nouvelles données et modèles prédictifs. Partie 2 : énergie de solvatation de Gibbs), Fluid Phase Equilib. 470 (2018) 91–100.
>> https://www.sciencedirect.com/science/article/pii/S0378381218301420
[7] C.-G. Pereira, L. Grandjean, S. Betoulle, N. Ferrando, C. Fejean, R. Lugo, J.-C.-de Hemptinne, P. Mougin, Phase equilibria of systems containing aromatic oxygenated compounds with CH4, CO2, H2, H2S, CO and NH3: Experimental data and predictions (Équilibres de phase des systèmes contenant des composés oxygénés aromatiques avec CH4, CO2, H2, H2S, CO et NH3 : Données expérimentales et prédictions), Fluid Phase Equilib. 382 (2014) 219–234.
>> http://www.sciencedirect.com/science/article/pii/S0378381214004567
[8] T.-K.-H. Trinh, J.-P. Passarello, J.-C. de Hemptinne, R. Lugo, Use of a non additive GC-PPC-SAFT equation of state to model hydrogen solubility in oxygenated organic compounds (Utilisation d'une équation d'état GC-PPC-SAFT non additive pour modéliser la solubilité de l'hydrogène dans des composés organiques oxygénés), Fluid Phase Equilib. 429 (2016) 177–195.
>> http://www.sciencedirect.com/science/article/pii/S0378381216303776
[9] T.-K.-H. Trinh, J.-C. de Hemptinne, R. Lugo, N. Ferrando, J.-P. Passarello, Hydrogen Solubility in Hydrocarbon and Oxygenated Organic Compounds (Solubilité de l'hydrogène dans les composés organiques hydrocarbonés et oxygénés), J. Chem. Eng. Data 61 (2016) 19–34.
>> https://pubs.acs.org/doi/full/10.1021/acs.jced.5b00119
Contact scientifique : Jean-Charles De Hemptinne
Vous serez aussi intéressé par

Nouvelle chaire dédiée à la thermodynamique des électrolytes
