




Maîtriser la compatibilité entre polymères et fluides est essentiel dans de nombreux secteurs de l’industrie, comme par exemple dans l’automobile avec la question de la tenue des matériaux du système d’alimentation en carburant. Pour les composants concernés, des méthodes expérimentales permettent d’anticiper d’éventuelles dégradations des propriétés initiales. Elles restent cependant très coûteuses en temps. Une approche alternative s’est avérée pertinente : utiliser des données existantes, en les complétant si nécessaire, pour en extraire de l’information via des méthodes issues de la science des données [1,2].
La chémoinformatique se situe à l’interface de plusieurs domaines scientifiques et consiste à utiliser des ressources informatiques pour résoudre des problèmes liés à la chimie. Une de ces déclinaisons est l’utilisation de l’intelligence artificielle pour prédire des propriétés d’usage. Outre la qualité des données, une des clés du succès réside alors dans la représentation des fluides complexes considérés, comme dans le cas des milliers de composés présents dans les carburants.
Malgré les avancées constantes des techniques d’analyse, identifier précisément chacun des constituants de ces fluides complexes paraît une tâche insurmontable. Grâce à la chromatographie bidimensionnelle (GC×GC), il est cependant possible de quantifier les familles chimiques présentes et d’obtenir des distributions en fonction du nombre d’atomes de carbone. A chaque composante de la distribution est associée une molécule représentative : à titre d’exemple, un carburéacteura peut ainsi être représenté structurellement par un mélange d’une centaine de composés [1].
Pour la rendre interprétable par des algorithmes d’apprentissage automatique, chacune de ces molécules doit être encodée par des descripteurs moléculaires. Comme il en existe un grand nombre, le choix s’est porté sur des descripteurs simples issus de dénombrements des groupes fonctionnels présents. Un vecteur descripteur encodant le fluide (figure 1) est ensuite obtenu grâce à une loi de mélange linéaire appliquée aux descripteurs, aux constituants et à leurs fractions respectives.
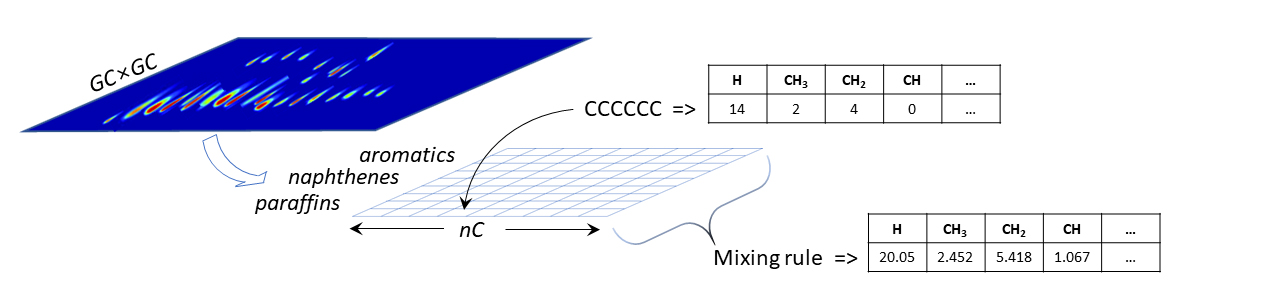
Cette méthodologie a été utilisée pour modéliser la quantité de fluide pénétrant dans un polymère lorsque ceux-ci sont mis en contact. Différents couples polymère/fluide ont ainsi été étudiés, des bases de données créées et des algorithmes d’apprentissage automatique appliqués.
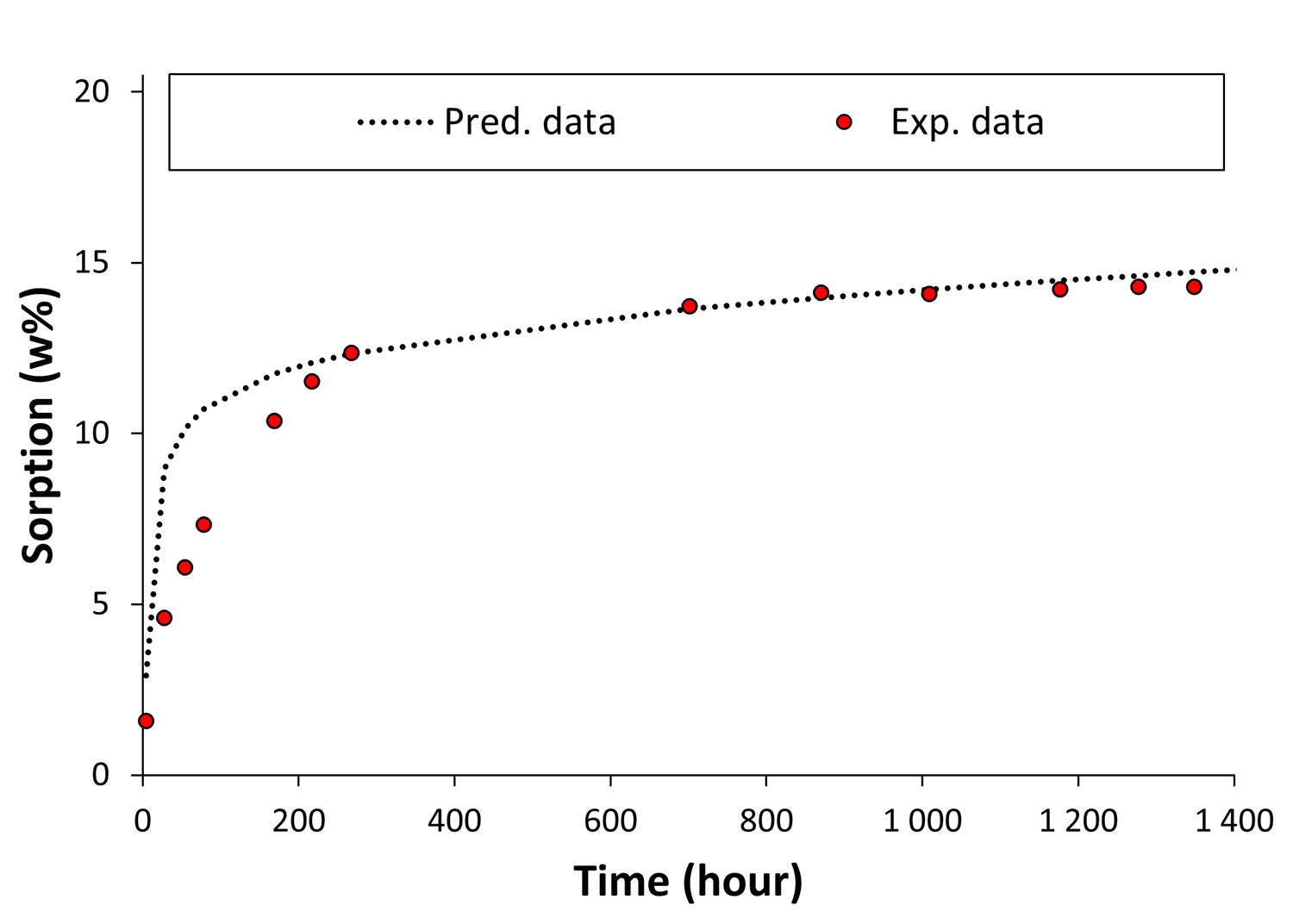
Les modèles obtenus permettent d’anticiper instantanément l’impact sur les propriétés de divers polymères de l’ajout de biocarburants dans les essences, ou encore de l’utilisation de carburéacteurs alternatifs (figure 2).
Une telle approche de modélisation contribue à réduire drastiquement le temps nécessaire pour quantifier la compatibilité polymère/fluide.
Dans ce contexte de la représentation des fluides complexes, au moins deux types de développements se dessinent :
- l’intégration de la génération in silico de structures [3] pour affiner la caractérisation ;
- l’application de cette méthodologie de modélisation à d’autres phénomènes, comme par exemple le vieillissement des fluides.
a- ou kérosène
b- matériau constitutif des circuits de carburant : réservoirs, tuyaux, joints
Références :
- B. Creton, B. Veyrat, M.-H. Klopffer, Fuel sorption into polymers: experimental and machine learning studies, Fluid Phase Equilibria 2022, 556, 113403.
>> DOI: 10.1016/j.fluid.2022.113403
- N. Villanueva, B. Flaconnèche, B. Creton, Prediction of alternative gasoline sorption in a semicrystalline poly(ethylene), ACS Combinatorial Science 2015, 17(10), 631-640.
>> DOI: 10.1021/acscombsci.5b00094
- C. Hall, B. Creton, B. Rauch, U. Bauder, M. Aigner, Probabilistic Mean Quantitative Structure Property Relationship modelling of Jet Fuels Properties, Energy & Fuels, 2022, 36(1), 463-479.
>> DOI: 10.1021/acs.energyfuels.1c03334
Contact scientifique : benoit.creton@ifpen.fr
Vous serez aussi intéressé par
La chémoinformatique et ses descripteurs : application à la compatibilité polymères/fluides
Maîtriser la compatibilité entre polymères et fluides est essentiel dans de nombreux secteurs de l’industrie, comme par exemple dans l’automobile avec la question de la tenue des matériaux du système d’alimentation en carburant.

Retour sur une chaire en thermodynamique pour les carburants issus de la biomasse
À la différence des molécules d’hydrocarbures d’origine fossile, celles issues de la biomasse sont polaires, en raison des hétéro-atomes qu’elles renferment. Cette différence à l’échelle moléculaire induit un comportement macroscopique plus complexe dont il faut tenir compte pour le dimensionnement des procédés qui les mettent en œuvre.
