




Tout le monde a remarqué que les batteries à base d’ions lithium, utilisées dans nos téléphones portables, ordinateurs, etc., perdent progressivement de l’autonomie au point de devenir inutilisables. Cette perte d’autonomie résulte notamment d’une couche, dite SEI1, qui s’installe entre une des électrodes et l’électrolyte de la batterie (cf. Figure). Cette couche apparaît dès le premier cycle de charge/décharge de la batterie, et croît progressivement en consommant des ions de lithium, de manière irréversible et donc au détriment de la capacité de la batterie [1].
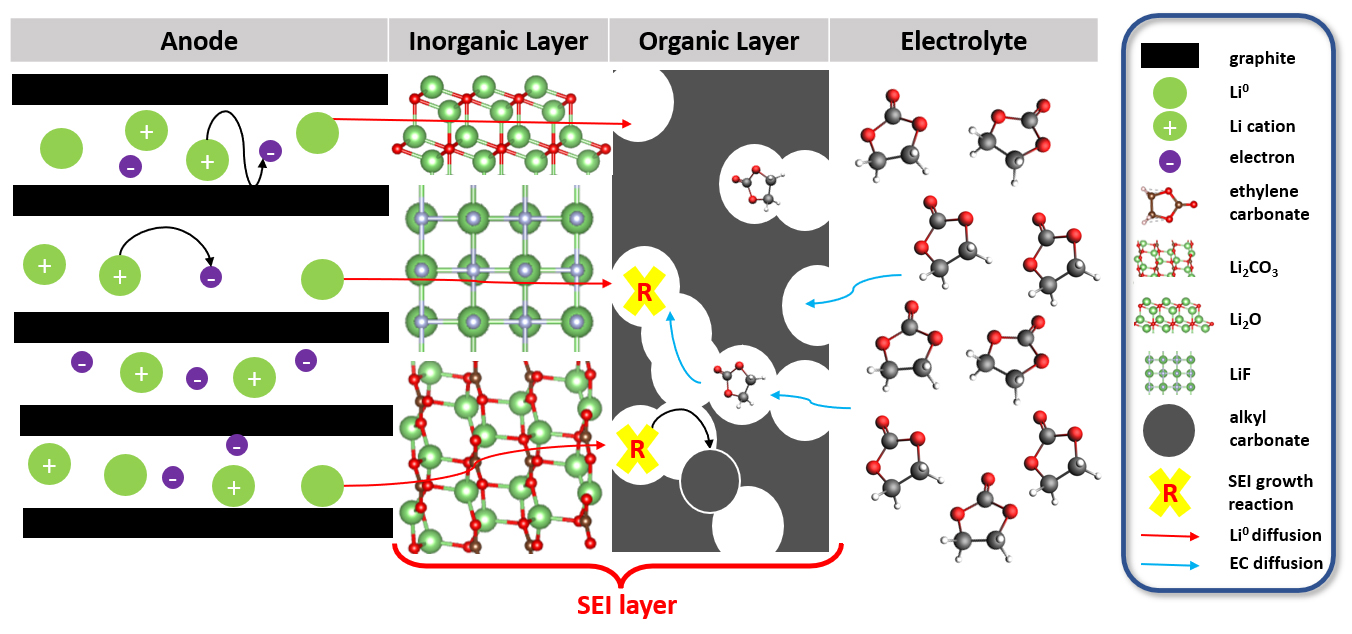
Si la composition chimique de cette couche SEI est aisément déterminée par des moyens expérimentaux d’analyse, d’autres aspects concernant sa formation, comme les conditions thermodynamiques de son apparition et sa cinétique de croissance, sont plus avantageusement étudiés par des approches théoriques, comme la modélisation moléculaire. Sur ce type de phénomène, la prise en compte des électrons est essentielle, car ils sont impliqués dans la réaction de réduction du solvant (composé de l’électrolyte) à l’interface de l’électrode négative, et c’est cette réaction qui initie et alimente la formation de la couche SEI.
Les méthodes basées sur la chimie quantique sont bien adaptées à ce besoin, car elles prennent en compte explicitement les électrons. Toutefois, elles sont trop consommatrices de ressources informatiques pour être appliquées à un système de la taille d’une batterie. Une approche multi-échelle apparaît dès lors comme une alternative pertinente, en permettant une description fine des réactions chimiques de dégradation du solvant sur des temps allant de la picoseconde jusqu’à la seconde, voire plus, et qui peut être étendue par le recours à la méthode de Monte Carlo cinétique (kMC2).
La première étape de ce travail de thèse a consisté à établir une base de données des réactions chimiques mises en jeu, avec leurs énergies de réaction et leurs énergies d’activation [2], grandeurs qui influent directement sur la vitesse de réaction. Ces énergies ont été calculées avec des méthodes basées sur la théorie de la fonctionnelle de la densité (DFT3).
Les simulations kMC réalisées ensuite ont notamment montré que des sels comme le Li2CO3 ou Li2O, issus de la dégradation de l’électrolyte, jouaient un rôle important dans la formation de la SEI [2]. Ces sels favorisent en effet l’apparition de diverses espèces organiques de dégradation, dont la présence a été corroborée par des observations analytiques, et qui engendrent une structure de SEI à couches multiples.
Les simulations kMC ont également permis de prédire la perte de capacité de la batterie en fonction de la composition de la couche SEI initialement formée (Figure 2). Il s’avère ainsi qu’en l’absence de Li2CO3 ou Li2O, cette décroissance de capacité est non-linéaire, et qu’en leur présence elle est plus lente. Ces résultats sont en accord avec d’autres études expérimentales et théoriques [3] et incitent à prendre aussi en compte l’apparition d’autres sels issus des réactions de dégradation, comme par exemple le LiF. Dans l’avenir, il est prévu d’élargir la bibliothèque de référence des réactions de dégradation pouvant se produire en présence de ces sels, et d’utiliser des méthodes moins gourmandes en CPU que la DFT.
1- Solid Electrolyte Interphase.
2- Méthode permettant de simuler le comportement de systèmes évoluant selon une équation maîtresse, en utilisant des données cinétiques des réactions chimiques élémentaires.
3- Density Functional Theory.
* Titre de la thèse : Mieux comprendre la formation et croissance de la Solid Electrolyte Interphase dans les batteries Li-ion par une approche de modélisation moléculaire.
Références :
-
A. Wang, S. Kadam, H. Li, S. Shi, Y. Qi, Review on modeling of the anode solid electrolyte interphase (SEI) for lithium-ion batteries, NPJ Comput. Mater. 4 (2018) 359.
>> https://doi.org/10.1038/s41524-018-0064-0
-
Mohammed Bin Jassar, Carine Michel, Sara Abada, Theodorus de Bruin, Sylvain Tant, Carlos Nieto-Draghi, Stephan N. Steinmann. A joint DFT-kMC study to model ethylene carbonate decomposition reactions: SEI formation, growth, and capacity loss during calendar aging of Li-metal batteries, ACS Appl. Energy Mater.
>> https://doi.org/10.1021/acsaem.3c00372
-
Kolzenberg, L. von; Latz, A.; Horstmann, B. Solid-Electrolyte Interphase During Battery Cycling: Theory of Growth Regimes. ChemSusChem 2020.
>> https://doi.org/10.1002/cssc.202000867
Contacts scientifiques : carlos.nieto@ifpen.fr ; theodorus.de-bruin@ifpen.fr
Vous serez aussi intéressé par

JIP EleTher : une communauté industrielle pour mieux comprendre les modèles thermodynamiques d'électrolytes
