

25.07.2024
3 minutes de lecture



Le calcul ab initio consiste à résoudre l’équation de Schrödinger pour un ensemble d’atomes représentant un système chimique d’intérêt. En réactivité chimique, la dynamique moléculaire ab initio (AIMD) permet par exemple de prédire les constantes de vitesse avec une grande précision, comme notamment dans le cas de zéolithes comportant des protons comme sites actifs. Néanmoins, les conditions d’obtention de résultats ayant la précision requise peuvent entraîner des temps de calcul déraisonnables. Une méthode récente (Machine Learning Perturbation Theory) permet de contourner cet obstacle. Le cas d’application choisi est celui de l’isomérisation et du craquage d’alcènes dans des zéolithes à larges pores.
Perspectives et défis du calcul ab initio
Le calcul ab initio consiste à résoudre l’équation de Schrödinger pour un ensemble d’atomes représentant un système chimique d’intérêt. Cette résolution nécessite de faire appel à des approximations plus ou moins fortes. Plus ces approximations sont limitées1 et plus la précision du calcul est améliorée, mais le coût du calcul important.
Dans le domaine de la réactivité chimique, un enjeu important pour le calcul ab initio est de pouvoir prédire les constantes de vitesse avec une précision satisfaisante2. Pour certains systèmes catalytiques, tels que les zéolithes comportant des protons comme sites actifs, cela requiert à la fois l’usage d’un niveau de théorie élevé et un échantillonnage de multiples configurations, par exemple par dynamique moléculaire ab initio (AIMD)3. Chacun de ces deux aspects nécessite des ressources computationnelles très importantes au point que, pour des calculs menés au niveau RPA (Random Phase Approximation)4, les combiner devrait en principe conduire à une résolution numérique pouvant durer un millénaire environ.
1 On parle alors de « niveau de théorie élevé »
2 Précision dite chimique, avec une déviation inférieure à 5 kJ/mol par rapport à des données expérimentales de référence
3 La dynamique moléculaire consiste à intégrer en fonction du temps les équations du mouvement dans le cas d’un ensemble d’atomes, en prenant en compte l’effet de l’agitation thermique, avec un pas de temps choisi. Des trajectoires sont ainsi obtenues, qui permettent d’échantillonner de nombreuses configurations (plusieurs dizaines/centaines de milliers) du système.
4 L’approche RPA permet de résoudre l’équation de Schrödinger à un niveau de théorie élevée et d’estimer l’énergie de corrélation électronique (énergie d'interaction entre électrons lié à leur influence mutuelle).
« Machine Learning Perturbation Theory » : une alliée bienvenue
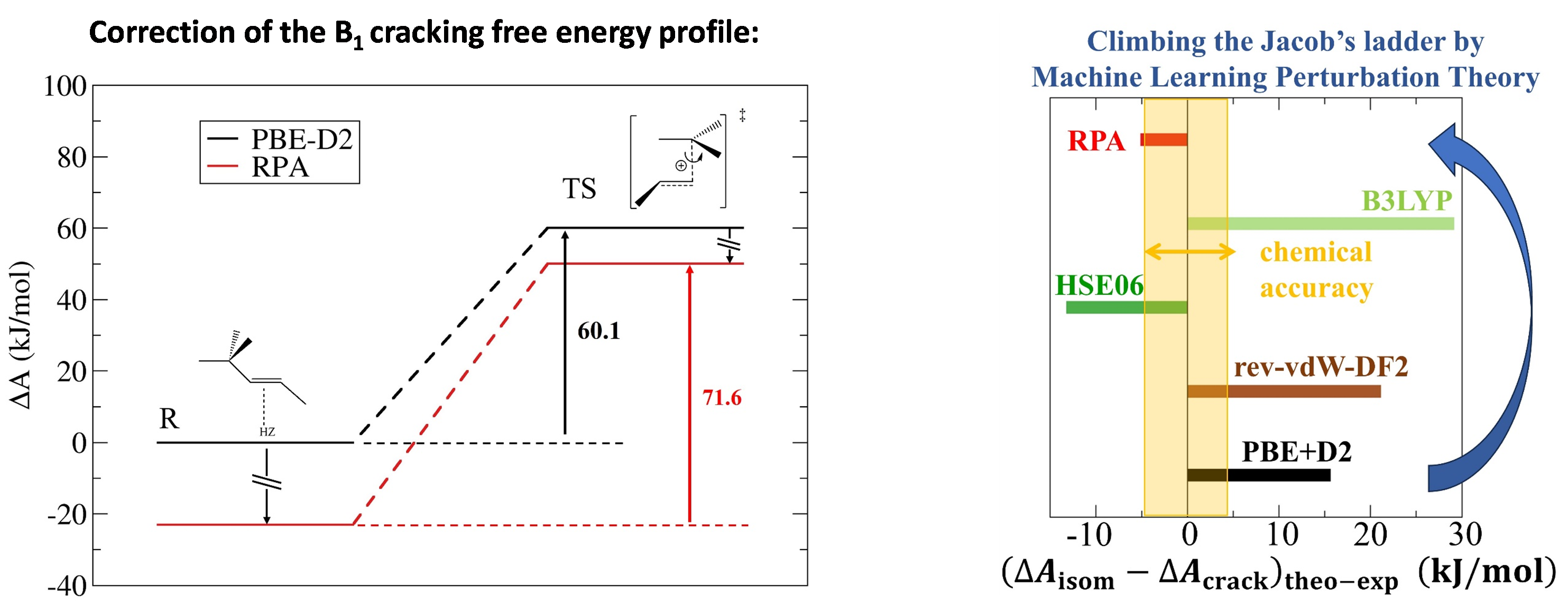
Cet obstacle de taille a été récemment contourné grâce à la mise en œuvre de la méthode MLPT (Machine Learning Perturbation Theory), développée à l’Université de Lorraine et à l’Université de Bratislava [1], et appliquée pour la première fois à un niveau de théorie aussi élevé [2], pour un calcul de constante de vitesse. Le cas de l’isomérisation et du craquage d’alcènes dans des zéolithes à larges pores (Figure 1, gauche) a été choisi dans la continuité de travaux précédents. Une accélération d’un facteur 4000 du calcul AIMD au niveau de théorie RPA a ainsi été atteinte. Par ailleurs, ce niveau est apparu nécessaire (Figure 1, droite) pour atteindre la précision attendue, par comparaison avec des données expérimentales acquises à IFPEN, et décryptées grâce à la modélisation cinétique [3].
Un futur usage tout trouvé : la transformation de molécules biosourcées
Ces travaux, également mis en lumière dans la revue Science [4], ouvrent la voie à la prédiction proche de la précision chimique de constantes de vitesse de réactions d’intérêt dans de nombreux domaines relevant des nouvelles technologies de l’énergie. C’est notamment le cas des réactions de transformation de molécules biosourcées, comme les alcools ou les sucres. Ainsi, des travaux récents menés en collaboration avec l’Université de Bratislava ont permis :
- de proposer les premiers mécanismes à même d’expliquer la formation de butènes linéaires à partir d’isobutanol [5] ;
- et plus récemment, de révéler par AIMD la complexité du réseau réactionnel [6].
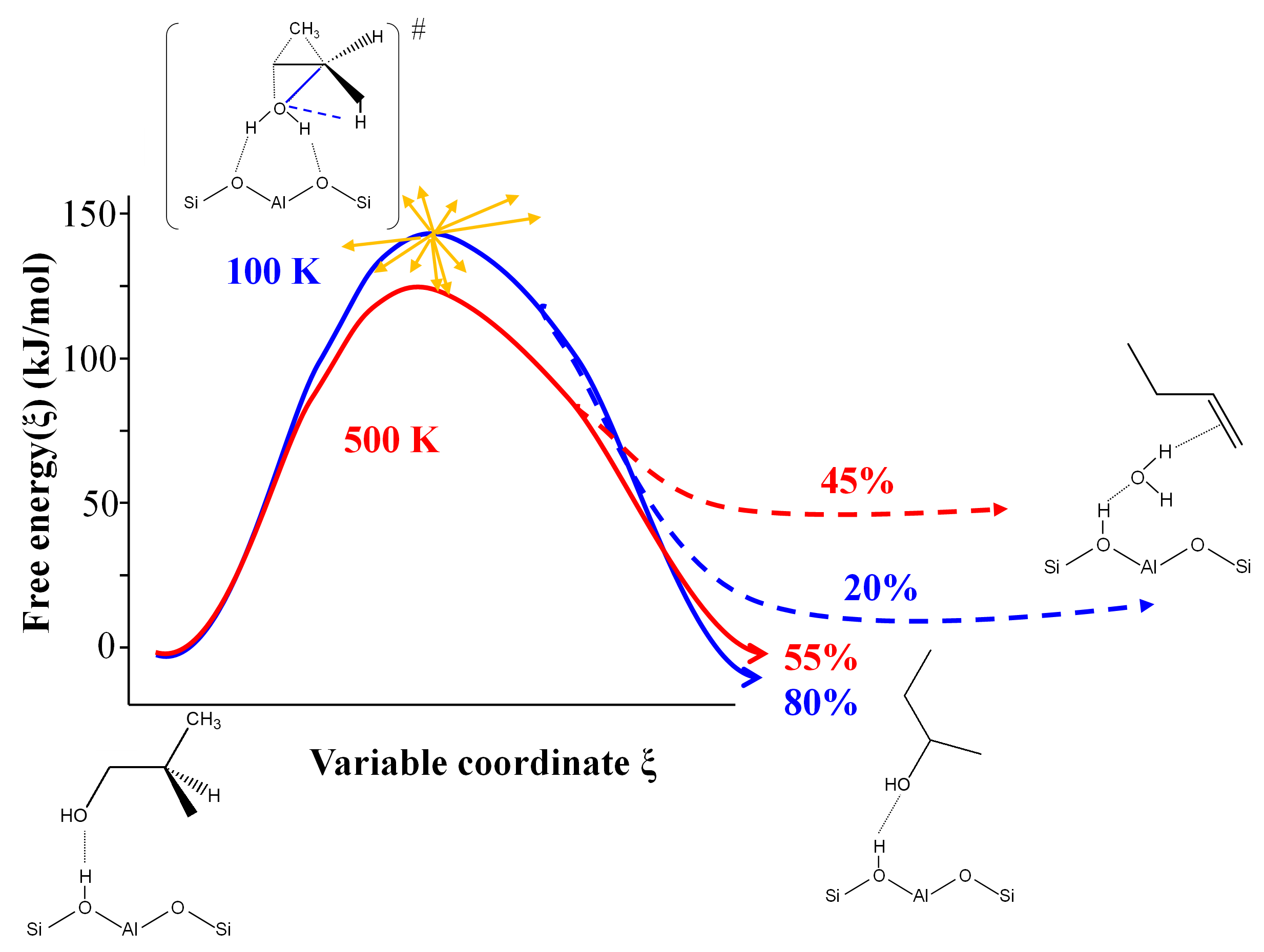
Dans ce dernier cas, le calcul AIMD démontre en effet que plusieurs réactions permettant la formation des butènes linéaires partagent le même état de transition, avec une bifurcation post-état de transition vers un produit ou un autre (Figure 2). Pour le calcul des constantes de vitesse, ceci empêche l’application traditionnelle de la théorie du complexe activé [7] et impose l’usage de méthodes AIMD avancées.
Le recours à la méthode MLPT est prévu dans le cadre du projet MAMABIO (PEPR B-BEST) pour améliorer la précision de ces données relatives à la déshydratation de l’isobutanol.
Références :
[1] Chehaibou, B.; Badawi, M.; Bucko, T.; Bazhirov, T.; Rocca, D., Computing RPA Adsorption Enthalpies by Machine Learning Thermodynamic Perturbation Theory, J. Chem. Theory Comput. 2019, 15, 6333-6342. http://dx.doi.org/10.1021/acs.jctc.9b00782
[2] Rey, J.; Chizallet, C.; Rocca, D.; Bučko, T.; Badawi, M., Reference-Quality Free Energy Barriers in Catalysis from Machine Learning Thermodynamic Perturbation Theory, Angew. Chem., Int. Ed 2024, 63, e202312392. https://doi.org/10.1002/anie.202312392
Rey, J.; Badawi, M., Rocca, D., Chizallet, C., Bučko, T, Machine learning thermodynamic perturbation theory offers accurate activation free energies at the RPA level for alkene isomerization in zeolites, Catal. Sci. Technol., 2024, 14, 5314-5323. https://doi.org/10.1039/D4CY00548A
[3] Schweitzer, J.-M.; Rey, J.; Bignaud, C.; Bučko, T.; Raybaud, P.; Moscovici-Mirande, M.; Portejoie, F.; James, C.; Bouchy, C.; Chizallet, C., Multiscale Modeling as a Tool for the Prediction of Catalytic Performances: The Case of n-Heptane Hydroconversion in a Large-Pore Zeolite, ACS Catal. 2022, 12, 1068-1081. https://doi.org/10.1021/acscatal.1c04707
[4] Ash, C.; Smith, J.; Jiang, D.; McCartney, M.; Ross, S. H.; Maroso, M.; Nusinovich, Y.; Suleymanov, Y.; Wible, B., In Other Journals, Science 2024, Vol 383, 380-381. https://doi.org/10.1126/science.ado2172
[5] Gešvandtnerová, M.; Bučko, T.; Raybaud, P.; Chizallet, C., Monomolecular mechanisms of isobutanol conversion to butenes catalyzed by acidic zeolites: Alcohol isomerization as a key to the production of linear butenes, J. Catal. 2022, 413, 786-802. https://doi.org/10.1016/j.jcat.2022.07.025
[6] Gešvandtnerová, M.; Raybaud, P.; Chizallet, C.; Bučko, T., Importance of Dynamic Effects in Isobutanol to Linear Butene Conversion Catalyzed by Acid Zeolites Assessed by AIMD, ACS Catal. 2024, 14, 7478-7491. https://doi.org/10.1021/acscatal.4c00736
[7] Eyring, H., The Activated Complex in Chemical Reactions, J. Chem. Phys. 1935, 3, 107-115. http://dx.doi.org/10.1063/1.1749604
Contact scientifique : Céline Chizallet
Vous serez aussi intéressé par
La dynamique réactionnelle dans les zéolithes à la loupe du calcul quantique
Les zéolithes sont des solides nanoporeux largement utilisés comme catalyseurs acides de transformation de molécules hydrocarbonées. Déterminer les vitesses des actes élémentaires des mécanismes réactionnels constitue toutefois un défi, en raison du grand nombre de degrés de liberté dont disposent les réactifs...
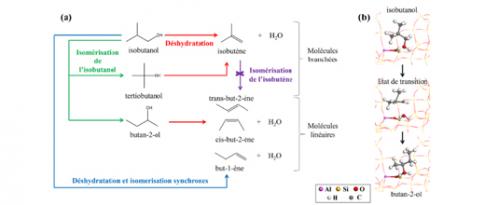
VS2 - Le calcul quantique révèle des mécanismes-clés pour la chimie biosourcée
La déshydratation d’alcools bio-sourcés en alcènes est une réaction-clé pour l’obtention de grands intermédiaires chimiques à partir de biomasse. Elle est efficacement catalysée par des zéolithes présentant des sites acides de Brønsted et un enjeu crucial est le contrôle de sa sélectivité...
Expériences et modélisation combinées pour étudier la transformation catalytique des sucres issus de biomasse
Motivé par l’enjeu mondial de basculer vers un modèle économique et énergétique plus durable, IFPEN s’intéresse depuis plusieurs années aux produits biosourcés à haute valeur ajoutée et travaille au développement de procédés de valorisation de la biomasse, comme une alternative à la pétrochimie classique.