




Thèse de Philippe Gantzer : « Développement et comparaison d’approches QSPR-inverse »
Quel ingénieur chimiste n’a jamais rêvé d’un outil lui permettant d’identifier directement un fluide (corps pur ou mélange) sur la base de caractéristiques nécessaires à un contexte applicatif donné ? Un tel Graal pourrait devenir réalité grâce à la Chémoinformatique et ses méthodes.
La Chémoinformatique se situe à l’interface de plusieurs domaines scientifiques et consiste à utiliser des ressources informatiques pour résoudre des problèmes dans le domaine de la chimie. Une de ses déclinaisons est l’utilisation de l’intelligence artificielle pour prédire des propriétés d’usage à partir de données de référence, relatives à la composition ou la structure [1]. Par l’apprentissage automatique, on peut ainsi établir des modèles qui sont autant de ponts entre des descripteurs moléculaires et des propriétés d’intérêt. Outre cette utilisation prédictive, la chémoinformatique peut être employée dans des processus de criblagea, voire s’inscrire dans une approche inverse, c’est-à-dire pour proposer des structures moléculaires susceptibles de satisfaire certaines contraintes.
Dans cette perspective, l’objectif plus particulier de ce travail de thèse a été de rechercher une méthode de génération de structures moléculaires. Après avoir recensé les différentes méthodes de la littérature [2], deux approches ont été retenues : la concaténation de fragments moléculaires (CFM) et l’évolution de structures moléculaires (ESM). La première consiste à identifier des fragments moléculaires puis à les combiner entre eux, tandis que la seconde consiste à faire évoluer des structures moléculaires par application de différents opérateurs, tels que des croisements ou des mutations d’atomes, de liaisons ou de groupes fonctionnels. Une fois implémentées sous forme numérique, ces approches ont été comparées entre elles selon une approche spécifique.
En effet, si il existe de nombreux critères pour évaluer la qualité prédictive d’un modèle décrivant des propriétés, il en existait peu pour les méthodes de génération de nouvelles structures. Pour pallier ce manque, nous avons proposé une démarche particulière dont la figure ci-dessous fournit une représentation graphique. Celle-ci consiste dans un premier temps à projeter les structures moléculaires virtuelles dans « l’espace chimique » – espace multidimensionnel fondé sur des descripteurs moléculairesb (et réduit ici à trois dimensions) – puis à comparer ces projections sur la base d’indices reflétant leur degré d’occupationc ou de couvertured de cet espace [3].
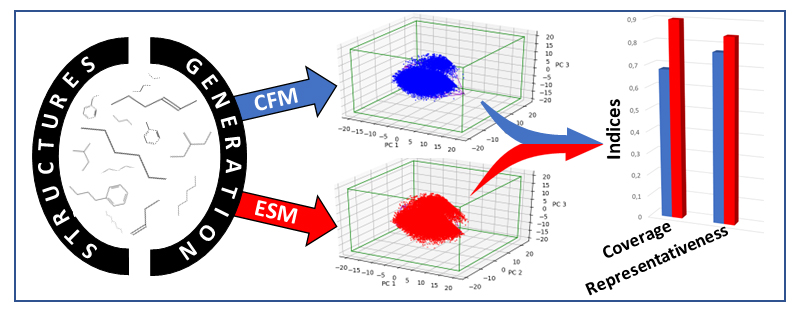
Les comparaisons réalisées sur la base de ces nouveaux indices ont montré que l’approche ESM est plus performante que la CFM. Elle permet de mieux couvrir l’espace chimique et est ainsi à même de proposer une plus grande diversité de nouvelles structures répondant à un cahier des charges donné.
C’est donc cette méthode qui va être utilisée dans différents contextes applicatifs, comme par exemple pour l’identification de nouveaux solvants.
a- Identification de molécules dans une base existante
b- Comme par exemple un dénombrement de groupes fonctionnels
c- Quantité de cubes élémentaires occupés
d- Peuplement individuel des cubes élémentaires
Références :
-
Science@ifpen numéro 48, juin 2022.
>> https://www.ifpenergiesnouvelles.fr/breve/chemoinformatique-et-ses-descripteurs-application-compatibilite-polymeresfluides
-
Gantzer, P.; Creton, B.; Nieto-Draghi C. "Inverse-QSPR for de novo Design: A Review", Molecular Informatics 2020, 39(4), 1900087.
>> https://doi.org/10.1002/minf.201900087
-
Gantzer, P.; Creton, B.; Nieto-Draghi C. "Comparisons of Molecular Structure Generation Methods Based on Fragment Assemblies and Genetic Graphs", Journal of Chemical Information and Modeling 2021, 61(9), 4245-4258.
>> https://doi.org/10.1021/acs.jcim.1c00803
Contacts scientifiques : benoit.creton@ifpen.fr ; carlos.nieto@ifpen.fr
Vous serez aussi intéressé par
Microfluidique et Chémoinformatique : une forte complémentarité pour étudier la compatibilité matériaux/fluides
Pour de nombreuses applications industrielles, comme le recyclage chimique des plastiques, ou encore pour assurer la compatibilité entre polymères et nouveaux carburants, il est essentiel d’anticiper les interactions entre matériaux et fluides...
La chémoinformatique et ses descripteurs : application à la compatibilité polymères/fluides
Maîtriser la compatibilité entre polymères et fluides est essentiel dans de nombreux secteurs de l’industrie, comme par exemple dans l’automobile avec la question de la tenue des matériaux du système d’alimentation en carburant.