




La chimie théorique au niveau quantique (théorie de la fonctionnelle de la densité, DFT) constitue un outil incontournable pour rationaliser les mécanismes réactionnels en jeu dans la préparation des catalyseurs ainsi que dans leur utilisation, grâce à une amélioration de leur activité [1,2]. De nombreux travaux d’IFPEN visent à élucider ces mécanismes catalytiques d’intérêt pour les procédés industriels [3,4,5] mais la simulation des étapes clés de la préparation des catalyseurs hétérogènes supportés a été peu abordée jusqu’alors.
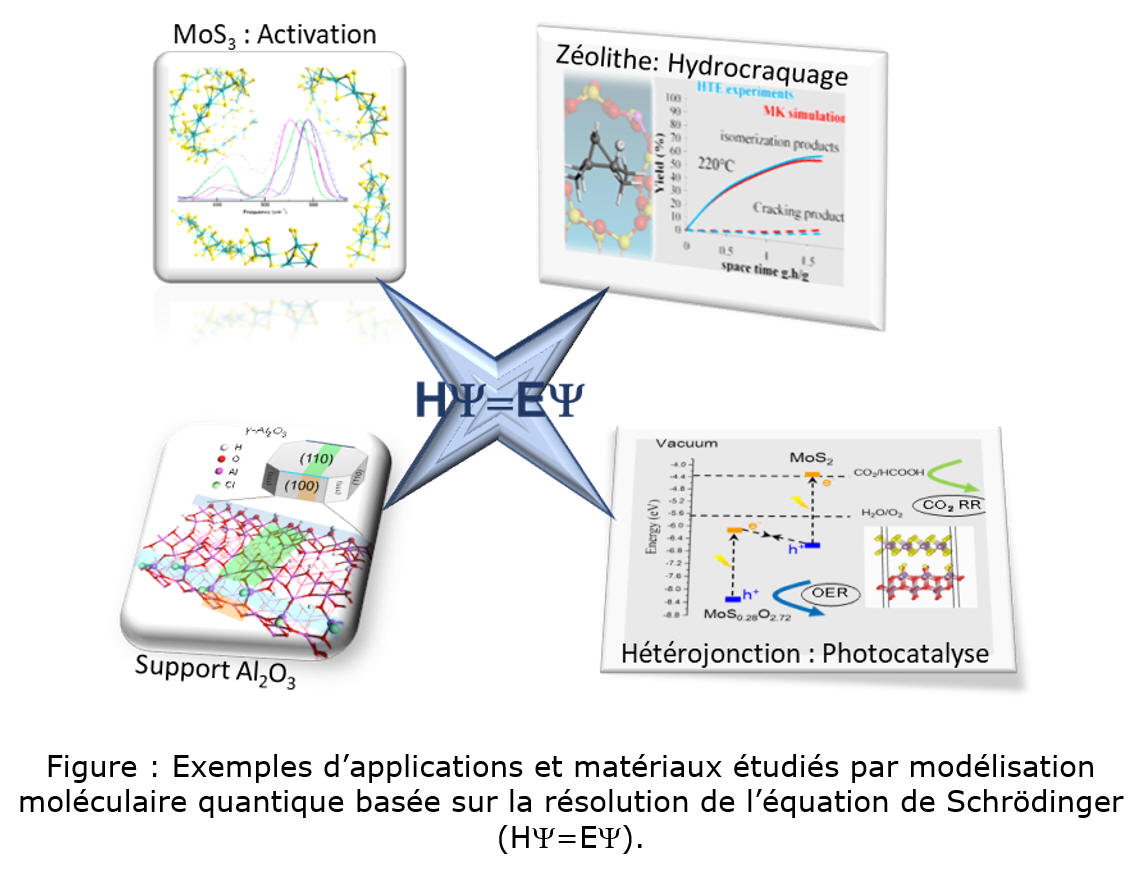
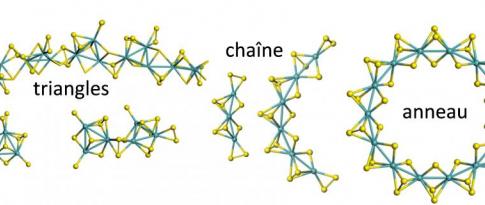
Les travaux de la chaire « Rational Design for Catalysis » (ROAD4CAT), menés en partenariat avec l’ENS de Lyon et l’Université Claude Bernard Lyon 1, ont entrepris de relever ce défi. Via une approche associant caractérisation par résonance magnétique nucléaire (RMN) et simulation par DFT, les travaux ont poussé plus loin la description des sites localisés sur le support d’alumine [6,7] et identifié les modes d’interaction d’additifs inorganiques phosphorés avec ce support [8]. Ces résultats fournissent des clés pour optimiser le dépôt des phases actives sur un support tel que l’alumine. Par ailleurs, concernant l’opération d’activation des catalyseurs, la simulation des mécanismes de transformation d’oligomères d’oxydes de molybdène en tri- et di-sulfures de molybdène (MoS3 et MoS2) a permis d’en identifier les étapes limitantes ainsi que les leviers d’amélioration [9].
Un second défi abordé par la chaire ROAD4CAT concerne la prédiction par des approches quantiques avancées des propriétés opto-électroniques de matériaux pour la photoréduction du CO2. L’identification d’hétérojonctions bidimensionnelles de type MoO3-xSx/MoS2 ou TiO2/MoS2 a ainsi ouvert des pistes d’identification de matériaux innovants pour orienter la séparation de charge (électrons-trous) via des processus « schéma en Z » inspirés de la photosynthèse [10].
Dans le cadre d’un projet de recherche fondamentale (projet Eyring), l’activité et la sélectivité en hydroconversion du n-heptane, catalysée par une zéolithe à large pore, ont pu être prédites grâce au couplage d’expérimentations à haut débit (EHD) et de modélisation microcinétique ab initio [11]. Au-delà du domaine du raffinage, une telle approche intéressera aussi potentiellement les procédés de recyclage des plastiques mettant en jeu des mécanismes analogues.
Un autre projet de recherche fondamentale est actuellement en cours pour répondre à l’enjeu méthodologique de l’amélioration du calcul des constantes cinétiques de réaction. Mené en partenariat avec l’Université Comenius de Bratislava, l’Université de Nancy Lorraine, l’Ecole des Ponts ParisTech et l’Inria, son approche consiste en la combinaison de la dynamique moléculaire quantique avec des algorithmes d’intelligence artificielle, de manière à accroître à la fois la vitesse et la précision du calcul de ces constantes.
Cliquer sur l'image pour l'agrandir
Production scientifique
Sur la période 2016-2021, sont parues environ 50 publications de la direction Catalyse, Biocatalyse et Séparation d’IFPEN associant de telles approches de modélisation moléculaire. Par ailleurs, les travaux des thèses de Kim Larmier et de Jérôme Rey ont été récompensés respectivement par les prix Yves Chauvin 2016 et 2020.
Références :
- M. Corral Valero, P. Raybaud, J. Catal. 391 (2020) 539.
>> https://doi.org/10.1016/j.jcat.2020.09.006
- C. Chizallet. Topics in Catal. (sous presse).
>> https://doi.org/10.1007/s11244-021-01489-y
- A. S. Dumon, A. Sahu, P. Raybaud. J. Catal. 403 (2021) 32.
>> https://doi.org/10.1016/j.jcat.2021.01.030
- W. Zhao, C. Chizallet, P. Sautet, P. Raybaud. J. Catal. 370 (2019) 118–129.
>> https://doi.org/10.1016/j.jcat.2018.12.004
- J. Rey, C. Bignaud, P. Raybaud, T. Bučko, C. Chizallet. Angew. Chem. Int. Ed. 132 (2020) 19100-19104.
>> https://doi.org/10.1002/ange.202006065
- A. T. F Batista, D. Wisser, T. Pigeon, D. Gajan, F. Diehl, M. Rivallan, L. Catita, A. S. Gay, A. Lesage, C. Chizallet, P. Raybaud. J. Catal. 378 (2019) 140.
>> https://doi.org/10.1016/j.jcat.2019.08.009
- T. Pigeon, C. Chizallet, P. Raybaud. J. Catal. 405 (2022) 140.
>> https://doi.org/10.1016/j.jcat.2021.11.011
- A. Hühn, D. Wisser, M. Corral Valero, T. Roy, M. Rivallan, L. Catita, A. Lesage, C. Michel, P. Raybaud. ACS Catal. 11 (2021) 11278.
>> https://doi.org/10.1021/acscatal.1c02135
- A. Sahu, S. N. Steinmann, P. Raybaud. Cryst. Growth Des. 20 (2020) 7750.
>> https://doi.org/10.1021/acs.cgd.0c00981
- M. Shahrokhi, P. Raybaud, T. Le Bahers, ACS Appl. Mater. Interfaces 13 (2021) 36465.
>> https://doi.org/10.1021/acsami.1c08200
- J.-M. Schweitzer, J. Rey, C. Bignaud, T. Bučko, P. Raybaud, M. Moscovici-Mirande, F. Portejoie, C. James, C. Bouchy, C. Chizallet. ACS Catal. (2022) 1068.
>> https://doi.org/10.1021/acscatal.1c04707
Contact scientifique : Pascal Raybaud
Vous serez aussi intéressé par

Méthodes de simulation moléculaire : mieux comprendre les premiers instants de la synthèse des zéolithes (en anglais)

Retour sur une chaire en thermodynamique pour les carburants issus de la biomasse
À la différence des molécules d’hydrocarbures d’origine fossile, celles issues de la biomasse sont polaires, en raison des hétéro-atomes qu’elles renferment. Cette différence à l’échelle moléculaire induit un comportement macroscopique plus complexe dont il faut tenir compte pour le dimensionnement des procédés qui les mettent en œuvre.
Un an d’existence pour la chaire ROAD4CAT
La 1re chaire industrielle au sein du projet d’IdexLyona, ROAD4CAT (RatiOnAl Design for CATalysis), associe depuis juin 2018 IFPEN et le laboratoire de Chimie