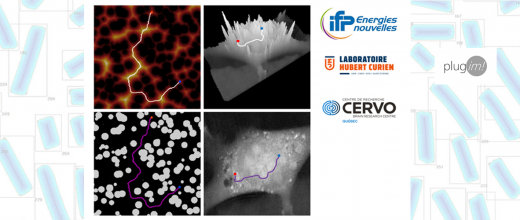
Dans les procédés catalytiques, une phase active est nécessaire pour accélérer la transformation des molécules du fluide traité. Cet agent catalytique est la plupart du temps déposé sur un support poreux doté d’une surface interne importante, permettant d’accueillir un grand nombre de sites actifs dans un faible volume. Cependant, outre la quantité et l’activité de la phase active, les performances d’un catalyseur sont aussi déterminées par l’accessibilité des molécules du fluide à ces sites de réaction. Améliorer les performances du catalyseur passe donc par une bonne description de la diffusion moléculaire effective au sein du support poreux, ce qui nécessite de mieux connaître la topologie interne de ce dernier.
Caractériser la porosité d’un matériau est habituellement réalisé par une technique d’adsorption et de désorption d’azote, méthode qui ne fournit toutefois que des informations indirectes sur la topologie et l’architecture du support poreux. Une représentation in silico de ce matériau peut cependant être obtenue en créant un jumeau numérique à l’aide d’un réseau de pores. Cet équivalent numérique constitue alors un modèle d’un grand intérêt pour comprendre comment les propriétés texturales et la topologie du réseau influencent la diffusion des molécules à travers la porositéa.
Afin d’élaborer ce modèle, des algorithmes ont été développés pour créer un réseau tridimensionnel de pores cylindriques interconnectés, dont les caractéristiques sont obtenues par échantillonnage Monte Carlo de distributions paramétrées [1]. À partir du réseau ainsi créé, il est possible de simuler numériquement les courbes d’adsorption et de désorption d’azote ou de porosimétrie par intrusion de mercure en utilisant des algorithmes dédiés, basés sur la théorie de la percolation par invasion. L’ajustement des paramètres du modèle s’effectue ensuite par le calage de ces données simulées sur les mesures expérimentales, ce qui permet d’adapter le réseau de pores à la structure de différents solides réels.
La validation du jumeau numérique ainsi obtenu a été effectuée par le calcul de son facteur de tortuosité, à l’aide de simulations de diffusion d’un traceur dans le réseau de pores. La comparaison au facteur de tortuosité expérimental, mesuré par RMN à gradient de champ pulsé sur un échantillon physique, a permis de valider l’approche [2].
En résumé, la représentation détaillée d’une structure poreuse, que les outils numériques permettent d’obtenir en réexploitant des données expérimentales communément disponibles, permet d’en extraire à moindre coût les informations topologiques. Dans le cas d’un support de catalyseur, cela permet de reproduire les chemins tortueux que les molécules doivent suivre afin d’atteindre la phase active. L’utilisation de ce type de modèle permettra à terme d’orienter la recherche vers des matériaux optimisés du point de vue de leur porosité, à l’aide de simulations de diffusion-réaction pour différents systèmes réactionnels.

a- Ces travaux ont été conduits dans le cadre de la thèse de Gabriel Alejandro Ledezma Lopez intitulée « Suitable Representations of Gamma Alumina Porous Structures by Computational Modeling », dirigée par Christian Jallut de l’université Claude Bernard Lyon 1.
Références :
- G.A. Ledezma Lopez, J.J. Verstraete, L. Sorbier, D. Leinekugel-Le-Cocq, E. Jolimaitre, C. Jallut, Computational Characterization of a Pore Network Model by Using a Fast Nitrogen Porosimetry Simulation, Computer Aided Chemical Engineering, 2021, vol. 50, pp. 1111-1116.
>> DOI: 10.1016/B978-0-323-88506-5.50171-6
- G.A. Ledezma Lopez, J.J. Verstraete, L. Sorbier, A. Glowska, D. Leinekugel-Le-Cocq, E. Jolimaitre, C. Jallut, Generation of γ-Alumina Digital Twins Using a Nitrogen Porosimetry Simulation, Industrial & Engineering Chemistry Research, 2021, vol. 60, n° 46, pp. 16728-16738.
>> DOI: 10.1021/acs.iecr.1c02849
Contact scientifique : jan.verstraete@ifpen.fr
Vous serez aussi intéressé par
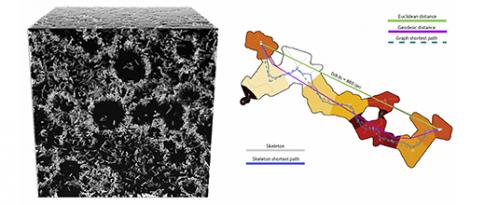
Conception numérique axée sur l’analyse de microstructures multi-échelles de matériaux poreux
La conception de matériaux poreux performants est un enjeu majeur pour l’efficience énergétique des procédés industriels : en catalyse, biocatalyse ou encore pour les opérations de séparation et de purification. Pour de telles applications, ces matériaux tirent leurs propriétés d’intérêt de leur microstructure particulière, comportant une grande quantité d’espaces vides organisés et connectés à l’échelle du nanomètre. IFPEN et Saint Gobain Research Provence (SGRP) se sont associés afin de se doter d’un outil facilitant à terme la mise au point de matériaux poreux optimisés en fonction d’usages donnés.
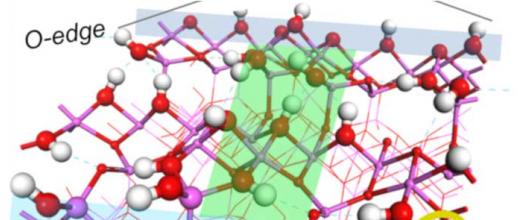
Spectroscopie et calcul quantique lèvent le voile sur les secrets des supports en alumine